|
Tetralogy of Fallot
The Tetralogy of Fallot defect consists of a combination of lesions that make up 10% of all congenital heart defects. It is the most common cyanotic heart defect with an incidence of 3.26 per 10,000 live births (Keane).
Anatomical description
The tetrad of lesions consists of: 1) stenosis of the pulmonary artery infundibulum; 2) a ventriculoseptal defect; 3) an overriding of the aortic root and valve; and 4) right ventricular hypertrophy. Tetralogy of Fallot has its developmental origins in a defective neural crest cell migration, which results in abnormal conotruncal formation. The four anatomic manifestations (above) result from the anterior malalignment of the infundibular septum.
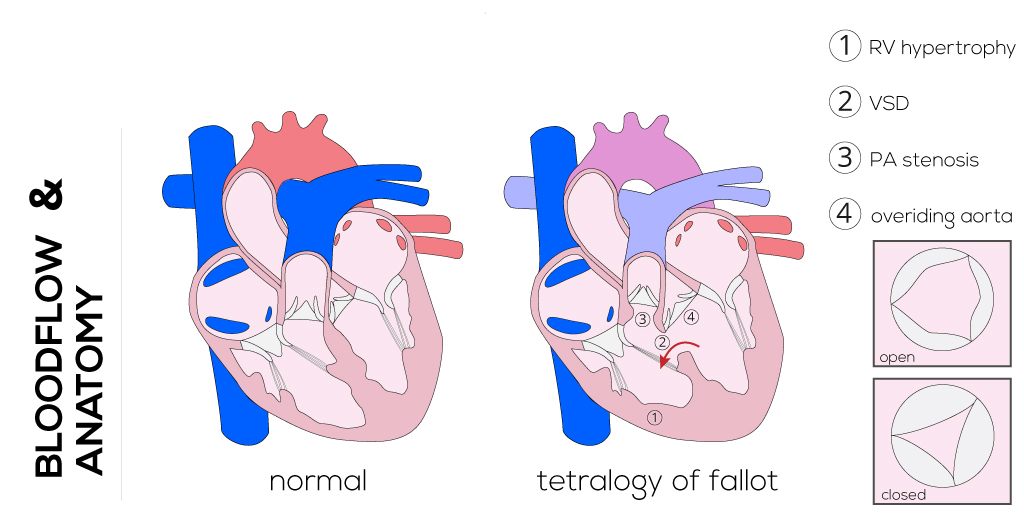
(Click picture to show/hide bloodflows)
Pathophysiology
Patients will present with cyanosis, known as blue babies, which is secondary to a greater degree of pulmonary and infundibular stenosis. This results in increased right-to-left shunting of blood across the VSD. The volume of this shunt and thus, the degree of cyanosis, is directly proportional to the severity of outflow obstruction. Shunting may also occur at the atrial level if there is an ASD or patent foramen ovale. In cases of Tetralogy of Fallot with pulmonary atresia, all pulmonary flow is derived from systemic-to-pulmonary communications (i.e., aortopulmonary collaterals, bronchial arteries, patent ductus arteriosus) which results in mixing of arterial and venous blood supplies. These left-to-right shunt communications place an increased volume load on the left ventricle which can lead to pulmonary hypertension. An absent or defective pulmonary valve (abnormal leaflets) leads to pulmonary regurgitation, and thus a volume load on the right ventricle and central pulmonary arteries. The right ventricle stroke volume increases to compensate for the volume load which leads to dilation of these arteries. The physiology of Tetralogy of Fallot with common atrioventricular canal is largely the same as tetralogy alone; however, in the case where the atrioventricular valve is insufficient, there may be increased volume load to one or both ventricles.
Infants with this defect will be variably cyanotic after birth, most often with only mild to moderate cyanosis; some may even be asymptomatic. Cyanosis typically increases in times of distress and with crying and hypercapnia. These hypercyanotic spells are a hallmark of Tetralogy of Fallot and are self-aggravating, whereby worsening hypoxemia exacerbates distress which then increases cyanosis. These episodes may often terminate in loss of consciousness and, rarely, convulsions and permanent neurological sequelae.
Therapy
The current recommendation for age of surgical repair is approximately 3 months, i.e., for infants born at full term and with uncomplicated anatomy. Repair during early infancy can restore normal circulation and minimize long-term risks associated with right ventricular hypertension and cyanosis.
Management of recurrent hypercyanotic spells that occur before surgery is urgent to prevent neurologic sequelae or even death. This may be done with supplemental oxygen and the administration of IV fluids, to increase intravascular blood volume and pulmonary blood flow. Medications such as morphine and propranolol have also been given to relieve distress, as well as beta adrenergic receptors.
Operative techniques including cannulation and myocardial preservation differ for various institutions and surgeons. Cardiopulmonary bypass, ventricular vent placement, aortic cross-clamping, and cardioplegia are usually applied for this procedure.
Palliative operations are designed to bridge the time until the infant reaches a certain age for the complete repair, such as the modified Blalock-Taussing Shunt procedure. Usually, a medial sternotomy is performed to create a shunt between the subclavian artery and the right or left pulmonary artery in order to provide a calibrated amount of pulmonary blood flow without using cardiopulmonary bypass. During the subsequent definitive operation, the shunt will be closed again.
Recently the transatrial approach has been adopted, aiming to correct the defect with one intervention. The treatment involves closure of the VSD via patch technique. Relief of the right ventricular outflow obstruction is achieved by resection of the obstructing infundibular muscle bands and right ventricular outflow patch reconstruction. In some cases it is required to extend across the annulus of the pulmonary valve to position the patch properly (transannular patch). After successful surgical correction, normal cardiac development of the chambers and regular physical capacity in adulthood is expected.
|