|
Single Ventricle
Anatomical description
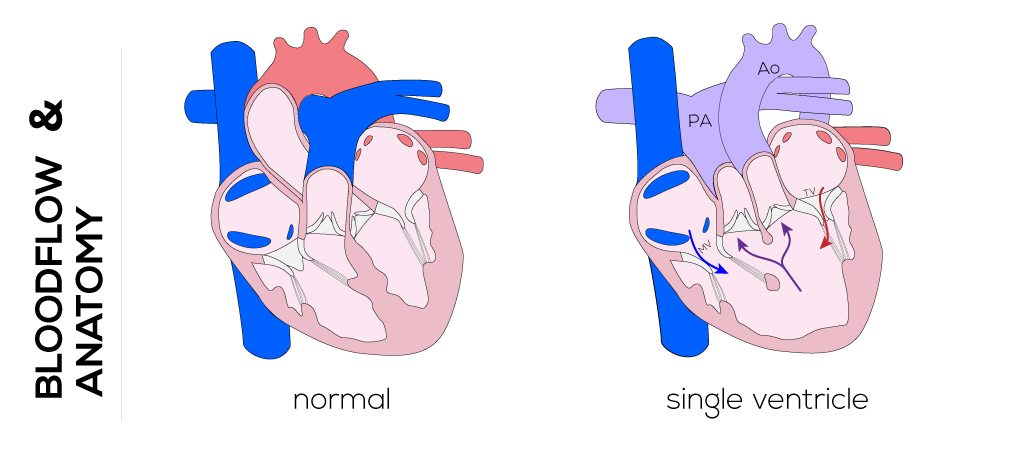
This defect describes a very rare congenital pathology. The single ventricle is characterized by the presence of two separate atrioventricular valves and either one ventricular chamber or a dominant ventricular chamber with diminutive opposing ventricle. The most common form of this defect is a single left ventricle with L-transposition of the great vessels. The aorta arises from a diminutive right ventricle that lies toward the left while the pulmonary artery arises more posteriorly .The tricuspid valve is found on the left side and mitral valve on the anatomical right side. This defect is often associated with ASD and further possible abnormalities in situ.
(Click picture to show/hide bloodflows)
Pathophysiology
The clinical course of infants with a single ventricle is determined by the amount of pulmonary blood flow limitation due to pulmonary stenosis or pulmonary vascular resistance. Without pulmonary stenosis, blood flow gradually increases to cause congestive heart failure, often within the first few days of birth. In the absence of stenosis, pulmonary vascular resistance cannot be reduced to normal levels and is ultimately incompatible with life. Infants with pulmonary stenosis show variable cyanosis at birth depending on the amount of blood flow via the ductus arteriosus, bronchial circulation, or aortopulmonary collaterals. In 80% of patients, blood from both the systemic and pulmonary circuits mixes in the single ventricle; thus, outflow through the aorta and pulmonary artery is of the same oxygen saturation. All patients with a single ventricle are cyanotic to some degree at birth.
Therapy
Initial management of the single ventricle defect focuses on lifesaving measures including administration of prostaglandins, placement of shunt, and/or catheterization. The ultimate goal is to perform a surgical Fontan procedure which establishes systemic circulation carrying fully saturated blood.
The first stage in surgical palliation is the Damus-Kayle-Stansel procedure. It aims to merge the proximal main pulmonary artery with the aorta above their respective valves to guarantee unobstructed aortic (systemic) blood flow. A homograft or autograft (pericardium) patch is used to augment the area where the two vessels are joined together. Regulation of the pulmonary blood flow can be managed by applying a Blalock-Taussing shunt which guarantees adequate perfusion of the lungs and prevents overcirculation by lowering the pulmonary pressure in comparison to the systemic pressure. This shunt is constructed by interposing a graft between the subclavian artery and the right or left pulmonary artery. Originally performed through a right/left thoracotomy depending on the neonate’s anatomy, the procedure is usually completed through a median sternotomy. It allows for a shorter shunt which can be placed more centrally.
In case of missing pulmonary stenosis, pulmonary banding may be applied to reduce the diameter of the artery and thus minimize pulmonary blood flow to prevent overcirculation of the lungs and pulmonary hypertension. The time of banding is limited due to somatic growth of the child and related cyanotic symptoms. At re-operation or second stage palliation, the band is removed and the vessel reconstructed.
The second stage is the bi-directional Glenn or Hemi-Fontan procedure performed on children in the following 2 to 10 months of life. The Glenn procedure is a superior cavopulmonary anastomosis as a first step to separate the systemic and pulmonary circulations, which reduces the volume work of the ‘single ventricle.. The Blalock-Taussing shunt is disconnected along with other additional sources of pulmonary blood flow. The superior vena cava is separated from the right atrium and connected to the right pulmonary artery. Consequently, deoxygenated blood from the head and neck flows directly into the pulmonary artery. The azygos vein is usually ligated too. The Hemi-Fontan is likewise a cavopulmonary anastomosis which is intended to simplify the eventual completion of the Fontan circulation. The superior vena cava is connected to the pulmonary artery while the interface between the superior vena cava and the right atrium is closed by a patch. An additional patch is used to endorse the merge of the superior vena cava with the pulmonary arteries and to address possible stenosis or distortion. When a lateral tunnel (versus an extra-cardiac conduit) is planned to accomplish total completion of the cavopulmonary circulation, the latter procedure is preferred.
Afterwards at the age of 18 to 24 months, the third stage and actual Fontan procedure will be performed. A pathway or tunnel either inside or outside of the heart (lateral atrial tunnel or extra-cardiac conduit) is constructed to connect the inferior vena cava with the pulmonary artery. A small opening in the venous pathway is created to compensate high right-sided pressures. Nearly normal oxygen saturation will be achieved after this procedure.
|